Rational
For Traditional Chinese Medicine (TCM) preparation research, DNA-based marker identification and downstream pharmacology analysis are in high demand. The advancement of related data accumulation and bioinformatics tools’ development has become the catalyst for great progress in this field. Until now, the majority of researches have been devoted to deciphering the TCM composition, as well as to mine TCM’s potential medicinal value. However, there is still a lack of an effective and accurate platform for TCM researches integrating composition identification and medicinal effect screening as a holistic pipeline.
Here, we developed the TCM-Suite platform that combined TCM identification with downstream network pharmacology investigation as a holistic platform. In TCM-Suite, two interconnected sub-databases named Holmes-Suite and Watson-Suite, were constructed for TCM composition identification and network pharmacology, respectively. In Holmes-Suite, we collected and pruned six existing marker gene (ITS2, matK, trnH-psbA,trnL, rpoc1, and ycf1) sequences for biological ingredient identification of TCM. After "Holmes" identified the species composition of TCM, "Watson" assisted in carrying out an in-depth network pharmacology investigation. In Watson-Suite, we curated heterogeneous types of records covering TCM herbs, gene targets, proteins and related diseases. Most importantly, using the machine learning method, a holistic pipeline connects the TCM composition identification and downstream network pharmacology analysis is constructed.
Content
Data collection and Process for Holmes-Suite and Watson-Suite
The data process for Holmes-Suite (exemplified by ITS2 marker gene)

Data source and quality filter of raw sequences
Raw marker gene sequencing data were extracted from the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/nucleotide/) in Genbank format searched with the words "marker gene", together with"Species" filtered to "Plants" in December 2019. The whole process workflow was exemplified by marker gene ITS2 (Figure 1), as below:
First, extractions of target ITS2 sequences of data downloaded were carried out with in-house scripts to trim boundaries of each ITS2 sequence, namely 5.8S and 28S rRNA genes which were highly conserved among different plant species (Figure 1A). Then, manual picking of sequences was performed to collect positive entries that the script didn’t cover. Due to the absence of ITS2 location annotations of some raw data, these sequences were moved to the candidate dataset first, and then a Hidden Markov Model (HMM) was trained based on well-annotated ITS2 sequences to predict the potential ITS2 regions of these candidate sequences before these ITS2 sequences could be included into our curated database. For all ITS2 sequences extracted based on the annotations, a quality filter was performed by criteria as follow (Figure 1A): (1) length below 100 bp, (2) length above 900 bp, (3) belonging to reduplicate entries, (4) with more than three ambiguous base pairs, (5) belonging to environment samples or unclassified samples. The quality control steps to filter ITS2 entries with either low sequence quality or obscure taxonomical annotation. Also, as there may be retrieval results with key words while containing no target sequence, which should be screened out.
Building and applying Hidden Markov Model
Due to the restriction of primers during amplification processes, ITS2 sequences obtained from the actual experiments usually contain sequences out of both boundaries, namely 5.8S and 28S rRNA genes in eukaryotes. With 1,000 sequences representing clean and complete ITS2 without ambiguous base pairs and out-of-boundary sequences from the data set passing the quality filter, a Hidden Markov Model of ITS2 sequences was trained through multiple sequence alignments with MUSCLE (Version 3.8.31) and HMMER3 (Version 3.1b2) to build the model with default parameters. The model was then applied to predict the boundaries of ITS2 regions of the candidate data set to extract target ITS2 sequences through the HMMER3 program. Through the search process based on probabilistic inference, those potential ITS2 sequences in the candidate data set could be extracted. These predicted sequences were also filtered in accordance with the criteria as set above. For sequences themselves or whose sub-sequences do not match the model, they were considered as non-ITS2 sequences and filtered out.
Metagenomic sample clustering approach to refine the ITS2 sequence database
Raw nucleotide sequences in NCBI might be in low quality: taxonomy information recorded of some sequences was not accurate or differed greatly of some sequences with high similarity. These problems would lead to deviation of organismal identification and taxonomic classification based on sequence similarity. To realize fault-tolerance and reduce the impact of the problems caused by original data, as well as to refine the database, sequence clustering approach used in metagenomics sample analysis (namely the UCLUST algorithm) was introduced (Figure 1B), which was generally used in a different context to generate clusters of (uncultivable or unknown) microorganisms (Operational Taxonomical Unit, OTU), grouped by DNA sequence similarity of a specific taxonomic marker gene. In the clustering process, sequences whose similarities above a certain value were grouped into a cluster expected to belong to the same species or closely related species.
The sequence cluster procedure was carried out by the UCLUST program (version v1.2.22q). Sequences were sorted through their length first and processed in order one by one. If a sequence being processed matched an existing centroid, it was assigned to that cluster, otherwise, it became the centroid of a new cluster. The similarity threshold of the ITS2 sequence was set to 0.99 so that highly homologous plant species could be clustered.
After the clusters were generated, we have performed further filtration for each cluster. Sequences whose phylogenetic relationships of species annotated diverging obviously in a cluster would be processed by the principle that isolated sequences (below 10% of the total sequences in the cluster) would be filtered while a dominant species in a majority number (above 90%) of the total sequences in the cluster would be retained. Finally, sequences with inaccurate annotations were filtered out.
Deployment and parameter setting of multiple search engines
As the taxonomic classification of applying the database was based on sequence similarity search, for the consideration of high accuracy and efficiency, multiple search engines were designed as Figure 1E. For alignment-based BLAST working in QIIME, data was formatted as two separated files containing sequences and taxonomy information, respectively. The mapping relation of ITS2 sequences and their species information was assigned. An efficient algorithm of sequence search, k-mer based Kraken, a fast and accurate algorithm initially used for assigning taxonomic labels to metagenomic DNA sequences, was also applied as an efficient species classification method. The core of Kraken was a database containing records consisting of a k-mer and the LCA (the least common ancestor) of all organisms whose genomes contain that k-mer. Sequences were classified by querying the database for each k-mer in a sequence and then using the resulting set of LCA taxa to determine an appropriate label for the sequence. As for Kraken, data was formatted (aligned to generate k-mers contained within the database used for Kraken) with built-in commands, and the NCBI taxonomy database was adopted as taxon information (mapped to k-mers with the GI number) for the construction of the custom Kraken database.
Network pharmacology data collection for Watson-Suite
Data sources for Watson-Suite
Watson-Suite was composed of five data fields, namely formula-herb, herb-ingredients, compounds, proteins, and diseases. In the current version, all the information and data were integrated based on related web-based databases.
Small molecular drug
For a small molecular drug, we could set a threshold of a combined score with the "compound-proteins/genes-disease" network obtained from our database. The information of the drug and its corresponding indications were collected from the Drugbank. Then we compared the indications with the disease from the network. If one of the diseases matched with recorded indications, we might find a candidate pathway for the drug to exert its effect.
The chemical constituents of Traditional Chinese Medicine were obtained from the TCM Database@Taiwan and TCMID. After obtaining all the processed compound files, we used the Can SMILE string or name of small molecules to search with the related genes and corresponding potential related-disease retrieved from our database. Unlike the small molecular drug, we took all the Traditional Chinese Medicine-related compounds as a whole. It relied on different compounds’ synergistic effects to exert its effect. Different compounds may act on the same pathway. Traditional Chinese medicine preparation. Traditional Chinese Medicine preparation consisted of several different Traditional Chinese Medicine.
Consistency consideration
The information of herb-ingredients such as herb and its related compounds with CID number were collected from TCMID. The information of compounds and their targets, diseases and their related proteins were obtained from STITCH and OMIM respectively. As the target ID used by STITCH and OMIM were different from each other, the data from these resources were inconsistent. To overcome the barriers, the information of proteins aliases and approved gene symbols were retrieved from OMIM to unite the data.
The protein interactions were obtained from STRING. The toxic and side-effect records of compounds were derived from TOXNET and SIDER. And the gene-disease associations were collected from GAD for facilitating the analysis.
Data integration and filtration, manual annotations and corrections
The compounds of ginseng and LDW that we downloaded from TCM Database@Taiwan were mol2 formats while the formats we used in STITCH were SMILES formats. To improve the compatibility of data, we converted all of them into a standard generalized format: Canonical SMILE format (Can). We used Open Babel to convert both mol2 and SMILE formats. into CAN formats. To further unify the format, the symbols represented configuration and chirality like "/", "\", "@", "[]" and "H" were removed from the CAN format in both the database and the herb-related compounds. With the unified format obtained, the Can SMILE string of compounds could be used to search with hits from the TCM-Mesh. We integrated data from various data sources, to make our data structure clearer, we further simplify our database. Firstly, the CID number was an internal connection between the compound and the protein, thus we could establish direct relations of compound and proteins. In addition, the data filed about herb-ingredient contained many compounds without SMILE string and CID number, which could not be used for further searching.
Secondly, target ID was different in various databases as we mentioned above. To unite our data, we used the protein aliases from OMIM to establish the relationship between the proteins and the genes, thus most of proteins with the corresponding genes of proteins have been retrieved from our database. The original table named protein aliases and approved gene were merged into one new table called “protein-gene links”. And protein interactions were also converted to the interactions of genes. We processed our data manually and removed the duplicate records.
Pipeline
Holmes
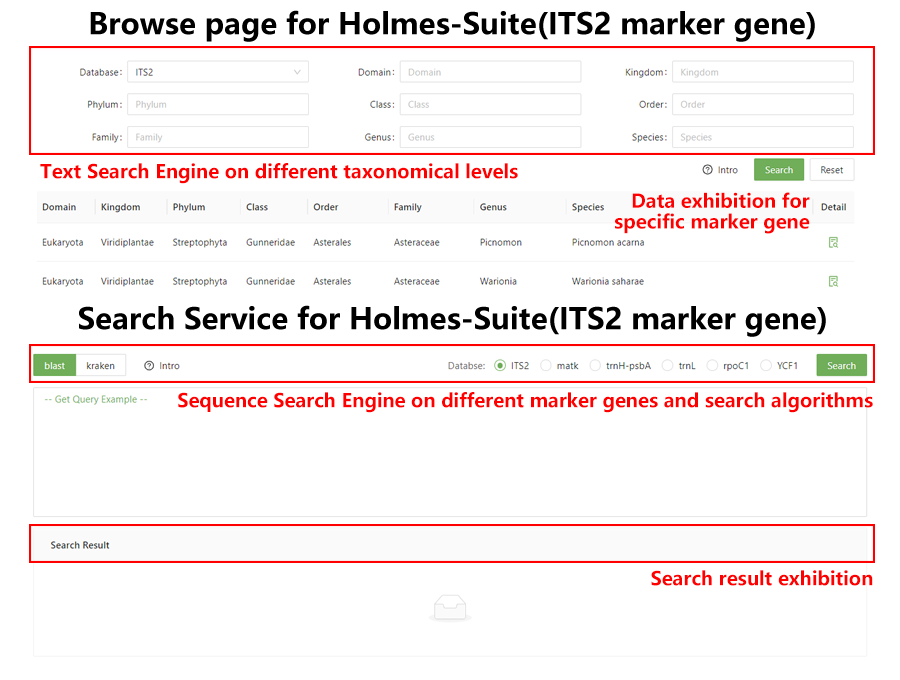 Figure 2. Screenshots of Holmes-Suite sub-database. The Holmes-Suite was composed by two main function modules: browse module (exhibit all the entries in Holmes-Suite) and search module (supplied the sequence search service for six marker genes).
Figure 2. Screenshots of Holmes-Suite sub-database. The Holmes-Suite was composed by two main function modules: browse module (exhibit all the entries in Holmes-Suite) and search module (supplied the sequence search service for six marker genes).The core function of the web service was organism identification and taxonomy classification of species based upon the ITS2 sequence. Namely, ITS2 sequencing data submitted and extracted by the HMM model was retrieved from the background database through the optional search engine and then species annotation was presented for each submitted sequence. Each batch of data submitted was treated as a sample, and the service also provided statistical information of the abundance of the species detected for the sample containing bulk data. Detailed species annotation results were displayed in a tabular form, supplemented by the statistical charts. The database also provided retrieval and browsing of the relevant original sequences and multiple query entries to ensure easy access to query, retrieve and filter information.
The main interface of the web service was shown in Figure 2 with main browse and search functional pages. In the browse page, all species sequences could be browsed, and their corresponding species’ phylogenetic positions and detailed descriptions were shown. On the search page, after submission of multiple sequences through the “Enter Query Sequence” window, selection of appropriate search engine by switching the label and corresponding DNA barcode dataset as background dataset, the species identification of the target sequences could be initiated.
Species identification results included the species information for each sequence displayed in a tabular form, and the species with top ten abundance of the sample at the taxonomy level of species and genus displayed in tables and pie charts, and showed the abundance composition of the entire sample (Figure 2).
Watson
 Figure 3. screenshot of Watson-Suite database. The Watson database supplied all the information for the network pharmacology. For each entry in Watson-Suite database, all the association information was listed in detailed page.
Figure 3. screenshot of Watson-Suite database. The Watson database supplied all the information for the network pharmacology. For each entry in Watson-Suite database, all the association information was listed in detailed page.Watson-Suite is designed as an integration of database and a data-mining system for network pharmacology analysis of TCM preparations (Figure 3). We have collected information on all respects of TCM including formula, herbs, herbal ingredients, targets, related disease, side effect and toxicity.
Users can browse, search and download the all the database entries through the web interface. Moreover, as an interconnect pipeline for TCM research, Watson-Suite also provided link to Holmes database for matched herbs, providing their marker genes.
Search
To search the information in Watson-Suite, users can firstly click the "Watson" button on the top right of the homepage, and then input and search for an interested query term in the search page. There are four different search boxes provided respectively. And for each search box, we provide different search types. For example, when users are searching herbs, three types of names (Pinyin name, Latin Name and English name) for the same herb are permitted to use (Figure 3).
Browse
After searching in Watson-Suite, matches for the input query terms are displayed in the lower part of the search page. The hyperlink is linked to the entry browse page which displays the detail information. Generally, detail page is designed in two parts: First, the Pharmacology network is displayed to display all the entries that identified to correlate with this entry in Watson-Suite. Second, the detailed information for all the entries is demonstrated by the order of Herb Information-Marker gene-Compound-Protein-Diseases (Figure 3).
Link to Holmes-Suite for marker gene of herbs
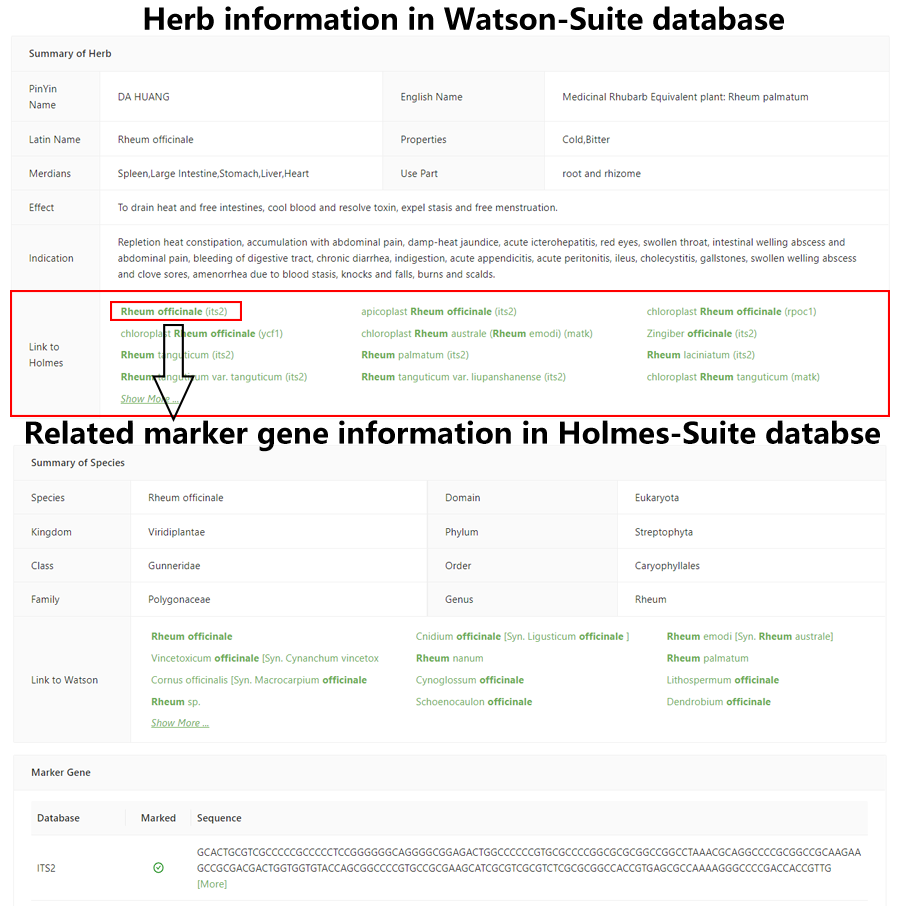 Figure 4. the link of Holmes and Watson-Suite database.
Figure 4. the link of Holmes and Watson-Suite database.We have realized the interconnection of the Watson-Suite and Holmes-Suite by connecting the "species" field in Holmes-Suite and the "herb" field in Watson-Suite (Figure 4), based on the fact that both stored the Latin name of biological composition of TCM. For these matched formulas, we integrated the detailed websites for biological composition of the two sub-databases: when users search for the biological composition of TCM in Holmes-Suite or determine the target herb in Watson-Suite, a link is provided for jumping to the corresponding database. Thus, a holistic pipeline for TCM research was born – from the identification of the biological composition of TCM to the exploration of related network pharmacology.
Examples
The coronavirus disease 2019 (COVID-19) has attracted great attention over the world since its explosion. Based on a large number of clinical practices, TCM was proven to play a significant role in the treatment of COVID-19, and was included in the guidelines on diagnosis and treatment of COVID-19. As a powerful platform for integrating TCM resources, using COVID-19-related network pharmacology constructed by the TCM-Suite platform, potential gene targets and therapeutic drugs could be mined for curing COVID-19's infection.
First, based on previous research, potential gene targets for curing COVID-19 were proposed and we constructed their potential pharmacological networks based on our database. We chose five target genes which were commended by previous researches for curing COVID-19. For these candidate genes, we constructed their "herb-gene/proteins-disease" networks using our database. For the five query genes (ACE2, SLC6A20, SIT1, CCR9, CXCR6), we identified compounds (12, 23, 1, 2, 14, respectively) and biological ingredients (58, 128, 2, 8, 54, respectively) for these target genes based on the entries in our database. Many records in our database were identified as associating with gene SLC6A20 and ACE2, which were also observed in previous researches. Further investigations into these potential target genes indicated that most of the effects of the 152 biological ingredients were "Clear heat and detoxify" and "Facilitate the flow of the lung and relieves toxicity", which were consistent with previous researches.
From another point of view, we investigated the clinical symptoms of COVID-19 infection to construct pharmacological networks. According to the clinical symptoms of COVID-19 infection referred from previous studies, we conducted a network pharmacological survey for these clinical symptoms (Table 1). Pneumonia is an important clinical symptom of COVID-19 infection, and interestingly, many records in our database associate with "Pneumonia".
| Disease | Gene target | Compound | Biological ingredient |
|---|---|---|---|
| SARS-coronavirus | 3 | 54 | 156 |
| Pneumonia | 10 | 67 | 138 |
| Typhoid fever | 8 | 60 | 215 |
| Cough | 5 | 46 | 215 |
| Dyspnea | 1 | 13 | 100 |
| Diarrhea | 12 | 25 | 125 |
Furthermore, we combined all these pharmacological networks, and obtained a target candidate gene cluster and potential therapeutic drugs for COVID-19. The common target genes and biological ingredients in these networks (occurred in over five pharmacological networks) were calculated, resulting in a total of 15 drug targets, 52 compounds and 86 biological ingredients. Many of these target genes have also been reported in previous researches, such as ACE2, SLC6A20, SIT1, CCR9, CXCR6, and we found that many of these 86 biological ingredients were identified as important components of clinically effective TCM to cure COVID-19. For example, biological ingredients Lonicera japonica,Scutellaria baicalensis, and Forsythia suspensa are the active ingredients of Shuanghuanglian; Forsythia suspensa, Ephedra sinica,Lonicera japonica, Isatis indigotica, Mentha haplocalyx, Dryopteris crassirhizoma, are the main component of Lianhua Qingwen capsule. All these TCM preparations were identified as effective drugs for curing COVID-19 infection. Based on this case study, The TCM-Suite platform has showed immense potential to shed light on the treasure house of TCM.
References
| 1. | Benson, D.A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Ostell, J., Pruitt, K.D. and Sayers, E.W. (2018) GenBank. Nucleic Acids Res, 46, D41-D47. |
| 2. | Eddy, S.R. (2009) A new generation of homology search tools based on probabilistic inference. Genome Inform, 23, 205-211. |
| 3. | Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res, 32, 1792-1797. |
| 4. | Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460-2461. |
| 5. | Ru, J., Li, P., Wang, J., Zhou, W., Li, B., Huang, C., Li, P., Guo, Z., Tao, W., Yang, Y. et al. (2014) TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform, 6, 13. |
| 6. | Sanderson, K. (2011) Databases aim to bridge the East-West divide of drug discovery. Nat Med, 17, 1531. |
| 7. | Zhang, R.Z., Yu, S.J., Bai, H. and Ning, K. (2017) TCM-Mesh: The database and analytical system for network pharmacology analysis for TCM preparations. Sci Rep, 7, 2821. |